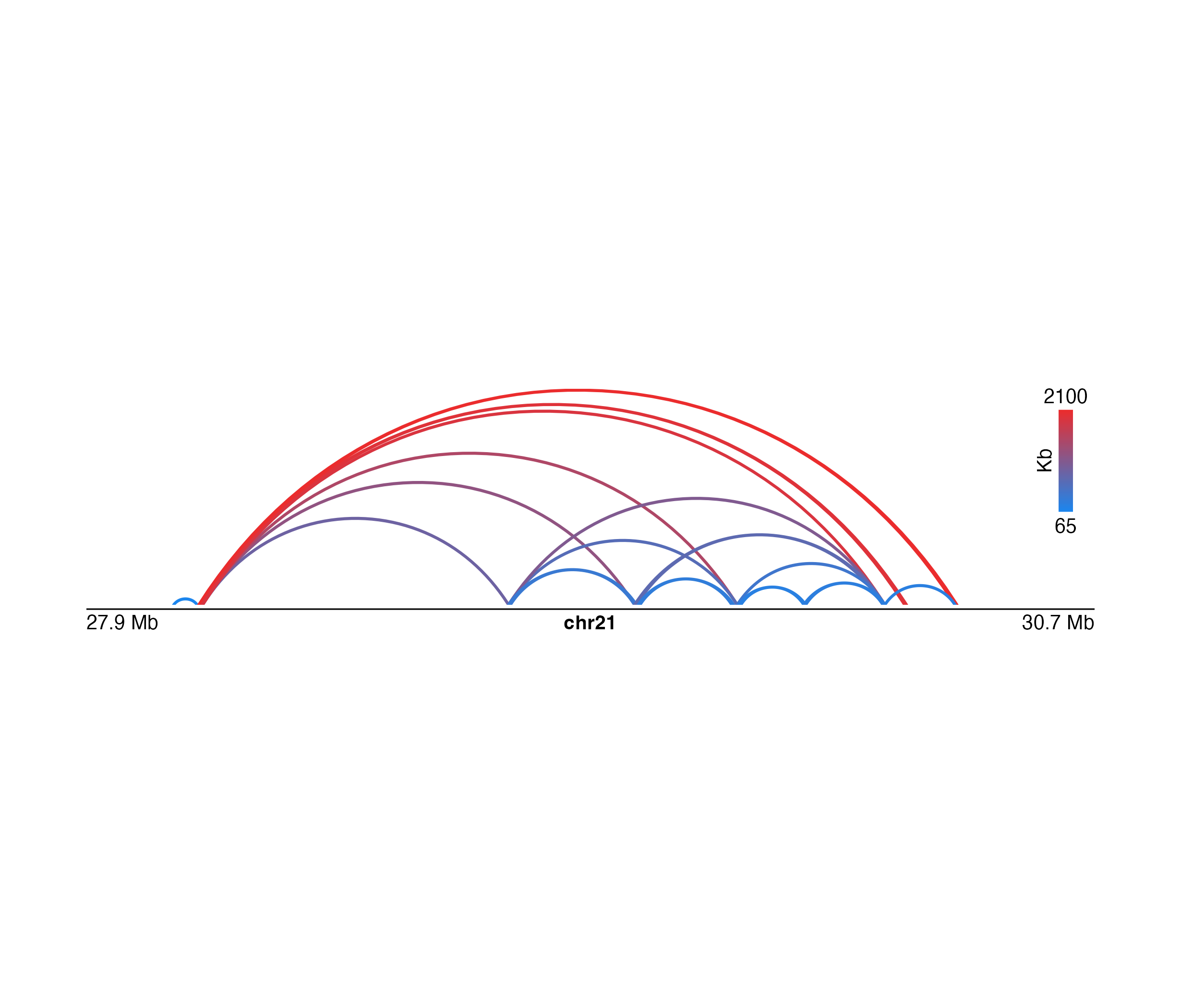
Plot paired-end genomic range data in an arch style
plotPairsArches(
data,
chrom,
chromstart = NULL,
chromend = NULL,
assembly = "hg38",
style = "2D",
flip = FALSE,
curvature = 5,
archHeight = NULL,
fill = "#1f4297",
linecolor = NA,
alpha = 0.4,
bg = NA,
clip = FALSE,
clip.noAnchor = TRUE,
range = NULL,
baseline = FALSE,
baseline.color = "grey",
baseline.lwd = 1,
x = NULL,
y = NULL,
width = NULL,
height = NULL,
just = c("left", "top"),
default.units = "inches",
draw = TRUE,
params = NULL,
...
)Arguments
- data
A string specifying the BEDPE file path, a dataframe in BEDPE format specifying data to be plotted, or a GInteractions object.
- chrom
Chromosome of region to be plotted, as a string.
- chromstart
Integer start position on chromosome to be plotted.
- chromend
Integer end position on chromosome to be plotted.
- assembly
Default genome assembly as a string or a assembly object. Default value is
assembly = "hg38".- style
Character value describing the style of arches. Default value is
style = "2D". Options are:"2D":Arches will be drawn in a 2-dimensional style.
"3D":Arches will be drawn in a 3-dimensional style.
- flip
Logical value indicating whether to reflect arches over the x-axis. Default value is
flip = FALSE.- curvature
Numeric indicating the number of points along the arch curvature. Default value is
curvature = 5.- archHeight
Single numeric value, numeric vector, or column name in data specifying the arch heights. When NULL, all arches will be the same height, filling up the given plot area.
- fill
A single character value, a vector, or a colorby object specifying fill colors of arches. Default value is
fill = #1f4297".- linecolor
A single character value, a vector, or a colorby object specifying the color of the lines outlining arches. Default value is
linecolor = NA. Special options include:NA:No line color.
"fill":Same color as
fill.
- alpha
Numeric value specifying transparency. Default value is
alpha = 0.4.- bg
Character value indicating background color. Default value is
bg = NA.- clip
A logical value indicating whether to clip any arches that get cutoff in the given genomic region. Default value is
clip = FALSE.- clip.noAnchor
A logical value indicating whether to clip any arches that overlap the given genomic region but do not have an anchor in that region. Default value is
clip.noAnchor = TRUE.- range
A numeric vector of length 2 specifying the y-range of
archHeightto plot (c(min, max)).- baseline
Logical value indicating whether to include a baseline along the x-axis. Default value is
baseline = FALSE.- baseline.color
Baseline color. Default value is
baseline.color = "grey".- baseline.lwd
Baseline line width. Default value is
baseline.lwd = 1.- x
A numeric or unit object specifying pair arches plot x-location.
- y
A numeric, unit object, or character containing a "b" combined with a numeric value specifying BEDPE arches plot y-location. The character value will place the pair arches plot y relative to the bottom of the most recently plotted plot according to the units of the plotgardener page.
- width
A numeric or unit object specifying pair arches plot width.
- height
A numeric or unit object specifying pair arches plot height.
- just
Justification of pair arches plot relative to its (x, y) location. If there are two values, the first value specifies horizontal justification and the second value specifies vertical justification. Possible string values are:
"left","right","centre","center","bottom", and"top". Default value isjust = c("left", "top").- default.units
A string indicating the default units to use if
x,y,width, orheightare only given as numerics. Default value isdefault.units = "inches".- draw
A logical value indicating whether graphics output should be produced. Default value is
draw = TRUE.- params
An optional pgParams object containing relevant function parameters.
- ...
Additional grid graphical parameters. See gpar.
Value
Returns a arches object containing relevant
genomic region, placement, and grob information.
Details
A pair arches plot can be placed on a plotgardener coordinate page by providing plot placement parameters:
plotPairsArches(data chrom,
chromstart = NULL, chromend = NULL,
x, y, width, height, just = c("left", "top"),
default.units = "inches")
This function can also be used to quickly plot an unannotated pair arches plot by ignoring plot placement parameters:
plotPairsArches(data, chrom,
chromstart = NULL, chromend = NULL)Examples
## Load paired ranges data in BEDPE format
library(plotgardenerData)
data("IMR90_DNAloops_pairs")
## Set the coordinates
params <- pgParams(
chrom = "chr21",
chromstart = 27900000, chromend = 30700000,
assembly = "hg19",
width = 7
)
## Create a page
pageCreate(width = 7.5, height = 2.1, default.units = "inches")
## Add a length column to color by
IMR90_DNAloops_pairs$length <-
(IMR90_DNAloops_pairs$start2 - IMR90_DNAloops_pairs$start1) / 1000
## Translate lengths into heights
IMR90_DNAloops_pairs$h <-
IMR90_DNAloops_pairs$length / max(IMR90_DNAloops_pairs$length)
## Plot the data
archPlot <- plotPairsArches(
data = IMR90_DNAloops_pairs, params = params,
fill = colorby("length", palette =
colorRampPalette(c("dodgerblue2", "firebrick2"))),
linecolor = "fill",
archHeight = "h", alpha = 1,
x = 0.25, y = 0.25, height = 1.5,
just = c("left", "top"),
default.units = "inches"
)
#> arches[arches1]
## Annotate genome label
annoGenomeLabel(plot = archPlot, x = 0.25, y = 1.78, scale = "Mb")
#> genomeLabel[genomeLabel1]
## Annotate heatmap legend
annoHeatmapLegend(
plot = archPlot, fontcolor = "black",
x = 7.0, y = 0.25,
width = 0.10, height = 1, fontsize = 10
)
#> heatmapLegend[heatmapLegend1]
## Add the heatmap legend title
plotText(
label = "Kb", rot = 90, x = 6.9, y = 0.75,
just = c("center", "center"),
fontsize = 10
)
 #> text[text1]
## Hide page guides
pageGuideHide()
#> text[text1]
## Hide page guides
pageGuideHide()