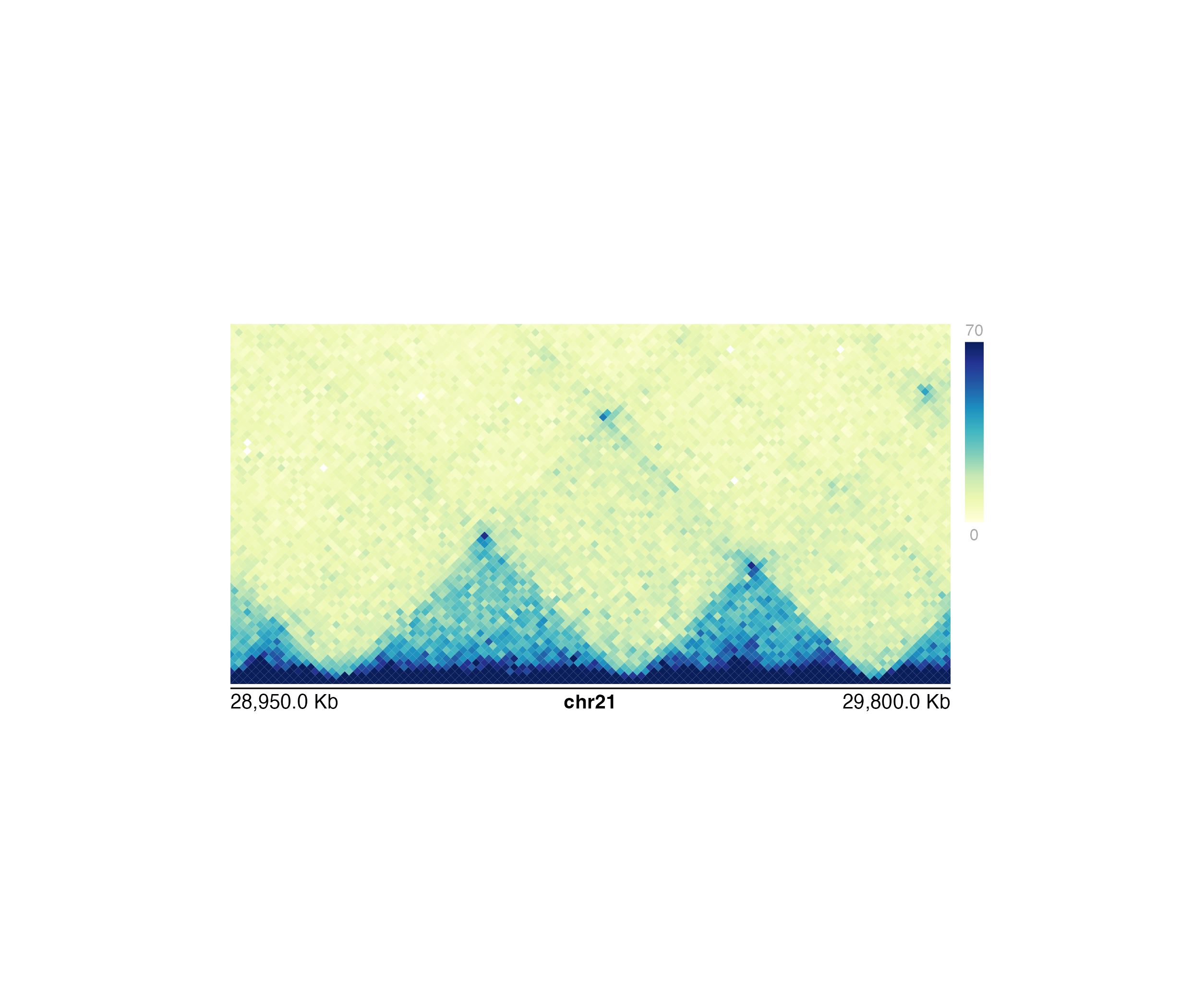
Plot a triangular Hi-C interaction matrix in a rectangular format
Source:R/plotHicRectangle.R
plotHicRectangle.RdPlot a triangular Hi-C interaction matrix in a rectangular format
plotHicRectangle(
data,
resolution = "auto",
zrange = NULL,
norm = "KR",
matrix = "observed",
chrom,
chromstart = NULL,
chromend = NULL,
assembly = "hg38",
palette = colorRampPalette(brewer.pal(n = 9, "YlGnBu")),
colorTrans = "linear",
flip = FALSE,
bg = NA,
x = NULL,
y = NULL,
width = NULL,
height = NULL,
just = c("left", "top"),
default.units = "inches",
draw = TRUE,
params = NULL,
quiet = FALSE
)Arguments
- data
Path to .hic or .(m)cool file as a string or a 3-column dataframe of interaction counts in sparse upper triangular format.
- resolution
A numeric specifying the width in basepairs of each pixel. For files, "auto" will attempt to choose a resolution based on the size of the region. For dataframes, "auto" will attempt to detect the resolution the dataframe contains.
- zrange
A numeric vector of length 2 specifying the range of interaction scores to plot, where extreme values will be set to the max or min.
- norm
Character value specifying hic data normalization method, if giving .hic or .(m)cool file. This value must be found in the .hic or .(m)cool file. Default value is
norm = "KR".- matrix
Character value indicating the type of matrix to output for .hic files. Default value is
matrix = "observed". Options are:"observed":Observed counts.
"oe":Observed/expected counts.
"log2oe":Log2 transformed observed/expected counts.
- chrom
Chromosome of region to be plotted, as a string.
- chromstart
Integer start position on chromosome to be plotted.
- chromend
Integer end position on chromosome to be plotted.
- assembly
Default genome assembly as a string or a assembly object. Default value is
assembly = "hg38".- palette
A function describing the color palette to use for representing scale of interaction scores. Default value is
palette = colorRampPalette(brewer.pal(n = 9, "YlGnBu")).- colorTrans
A string specifying how to scale Hi-C colors. Options are "linear", "log", "log2", or "log10". Default value is
colorTrans = "linear".- flip
A logical indicating whether to flip the orientation of the Hi-C matrix over the x-axis. Default value is
flip = FALSE.- bg
Character value indicating background color. Default value is
bg = NA.- x
A numeric or unit object specifying rectangle Hi-C plot x-location.
- y
A numeric, unit object, or character containing a "b" combined with a numeric value specifying rectangle Hi-C plot y-location. The character value will place the rectangle Hi-C plot y relative to the bottom of the most recently plotted plot according to the units of the plotgardener page.
- width
A numeric or unit object specifying the width of the Hi-C plot rectangle.
- height
A numeric or unit object specifying the height of the Hi-C plot rectangle.
- just
Justification of rectangle Hi-C plot relative to its (x, y) location. If there are two values, the first value specifies horizontal justification and the second value specifies vertical justification. Possible string values are:
"left","right","centre","center","bottom", and"top". Default value isjust = c("left", "top").- default.units
A string indicating the default units to use if
x,y,width, orheightare only given as numerics. Default value isdefault.units = "inches".- draw
A logical value indicating whether graphics output should be produced. Default value is
draw = TRUE.- params
An optional pgParams object containing relevant function parameters.
- quiet
A logical indicating whether or not to print messages.
Value
Returns a hicRectangle object containing
relevant genomic region, Hi-C data, placement,
and grob information.
Details
This function is similar is plotHicTriangle but will fill in additional pixels around the the triangular portion of the plot to make a rectangle. The x-axis represents the genomic coordinates and the y-axis corresponds to distance in Hi-C bins.
A rectangle Hi-C plot can be placed on a plotgardener coordinate page by providing plot placement parameters:
plotHicRectangle(data, chrom,
chromstart = NULL, chromend = NULL,
x, y, width, height, just = c("left", "top"),
default.units = "inches")This function can also be used to quickly plot an unannotated rectangle Hi-C plot by ignoring plot placement parameters:
plotHicRectangle(data, chrom,
chromstart = NULL, chromend = NULL)See also
Examples
## Load Hi-C data
library(plotgardenerData)
data("IMR90_HiC_10kb")
## Create a page
pageCreate(width = 6, height = 3.5, default.units = "inches")
## Plot and place rectangle Hi-C plot
hicPlot <- plotHicRectangle(
data = IMR90_HiC_10kb, resolution = 10000,
zrange = c(0, 70),
chrom = "chr21",
chromstart = 28950000, chromend = 29800000,
assembly = "hg19",
x = 0.5, y = 0.5, width = 5, height = 2.5,
just = c("left", "top"),
default.units = "inches"
)
#> Read in dataframe. Assuming 'chrom' in column1 and 'altchrom' in column2. 10000 BP resolution detected.
#> hicRectangle[hicRectangle1]
## Annotate x-axis genome label
annoGenomeLabel(
plot = hicPlot, scale = "Kb", x = 0.5, y = 3.03,
just = c("left", "top")
)
#> genomeLabel[genomeLabel1]
## Annotate heatmap legend
annoHeatmapLegend(
plot = hicPlot, x = 5.6, y = 0.5,
width = 0.13, height = 1.5,
just = c("left", "top")
)
 #> heatmapLegend[heatmapLegend1]
## Hide page guides
pageGuideHide()
#> heatmapLegend[heatmapLegend1]
## Hide page guides
pageGuideHide()